
A Unifying Grammar for DNA Variants
A unified framework and toolkit for standardizing DNA variants at scale. Part 1 of the neoantigen discovery with long-read sequencing series.

TL;DR
Current genomic variant representations are inconsistent across different variant callers and limit neoantigen discovery.
New unified variant grammar addresses the fundamental issue.
New tool VSTOL lets you standardize DNA variants from 14 different variant callers.
1. Short-Sighted in a Long Genome
We know that CD8+ killer T cells have the remarkable ability to selectively kill cancer cells in many cancer types. Immune checkpoint blockade therapies such as PD-1 inhibitors (e.g. Nivolumab and Pembrolizumab) and PD-L1 inhibitors (e.g. Atezolizumab and Avelumab) demonstrate this ability either by releasing the “brakes” on the killer T cells or by exposing the “disguise” of tumors cells to killer T cells. At the same time, we often don’t know which tumor-specific targets the killer T cells recognize to distinguish tumor cells (non-self) from normal cells (self). Occasionally, we discover tumor-specific peptide-major histocompatibility complex (pMHC) molecules that are recurrently presented across multiple patients with the same cancer type. For example, the cancer-testis antigen (CTA) NY-ESO-1 is expressed in over 70% of synovial sarcomas while PRAME, another CTA, is found in more than 80% of melanomas. These recurrently expressed CTAs have been the targets of effective adoptive T-cell receptor (TCR) therapies, as demonstrated by D’Angelo et al. and Wermke et al.
However, we also know that most cancers are characterized by both inter- and intra-patient heterogeneity rather than homogeneity. As a result, TCR therapeutics developed for shared targets remain limited to only a handful of cancer types. This challenge has driven significant efforts towards the development of personalized immunotherapies that individualize target selection for each cancer patient. Despite the promising scientific narrative behind this precision oncology approach, most personalized immunotherapies have been unsuccessful. Neon Therapeutics, Gritstone bio, and PACT Pharma are just some of the companies that have attempted but failed to deliver successful personalized immunotherapies.
There have been a few notable successes in personalized immunotherapy development. These successes are characterized by either hedging across many different target predictions or by directly using TCRs from antigen-experienced tumor-infiltrating lymphocytes.
So why does personalized immunotherapy usually fail? At PIRL, we think that at least one major deficiency is in the first few steps of the target selection process.
Target selection for personalized immunotherapy development relies on identification of neoantigens. Neoantigens are short (8-11 amino acids in length) mutant peptides presented by MHC class I molecules on tumor cells. The neoantigen identification process typically involves sequencing the tumor DNA and RNA as well as the matched normal DNA. The process begins by detecting somatic DNA variants, which are exclusively found in the genomes of tumor cells. This step ensures that personalized immunotherapies target tumor-specific molecular features and minimize the risk of autoimmune reactions against healthy and normal cells.
Over the past two decades, personalized immunotherapy development has suffered from the streetlight effect of looking at a specific type of somatic DNA variants: single-nucleotide variants (SNVs) that result in single-amino acid substitutions. As the name implies, these changes are subtle - so subtle that they might be indistinguishable from self-antigens on normal cells to killer T cells. If I wrote, “I hope you have a greit day,” you would still know what I meant - unless you speak Norwegian and know that greit means okay.
SNVs are typically detected in formalin-fixed paraffin-embedded (FFPE) tumor samples using short-read whole-exome sequencing - an assay that profiles only ~1% of the human genome (30 million out of the 3,000 million base pairs in the genome). By design, this target discovery setup overlooks the remaining 99% of the genome.
Could the most clinically actionable mutations lie in regions where we haven’t examined carefully? Could there be sources of therapeutic targets qualitatively better, less self-like, and more immunogenic? Could the answers we’ve been searching hidden in plain sight - just beyond the boundaries we’ve chosen to search?
As a field, we might have been short-sighted in a long genome.
2. Bigger Mutations, Bigger Impact?
So the natural question that arises is, “Are there bigger changes in tumor genomes?” The answer is yes. The Pan-cancer Whole Genomes Analysis Group (PCAWG) has revealed somatic structural variants (commonly defined in the literature as 30-50 base pairs or larger) in genomes of over 2,600 cancer patients representing 33 tumor types. There are 5 major types of structural variants: insertions, deletions, duplications, inversions, and translocations. If somatic structural variants affect the open-reading frames (ORFs) of protein-coding transcripts or otherwise give rise to novel ORFs, structural variants could represent a large pool of previously untapped neoantigens potentially highly divergent from the landscape of self-antigens. Given their inherently large sizes, somatic structural variants have the capacity to generate a bigger number of candidate mutant sequences subject to neoantigen prediction compared to SNVs (Figure 1). Also, tumors previously thought to have a low mutational burden (a metric typically based on the number of SNVs) might have targetable somatic structural variants. Moreover, the growing adoption of long-read sequencing means that we are better powered to identify and resolve somatic structural variants in cancer genomes.

In fact, a recent cancer vaccine trial has shown that frameshift mutations (insertions and deletions) elicited the highest in vitro (ELISPOT) immune responses in 4 out of the 9 patients that were enrolled in the study, and showed comparable responses to SNVs in 2 additional patients (see the publication’s Extended Data Figure 2b).
However, as you’ll see, targeting such mutations is difficult given the existing toolkits and ways of thinking about variants. So my PhD thesis work is focused on addressing this challenge and discovering neoantigens that originate from somatic structural variants in tumors.
3. One Variant, Multiple Representations
What is a DNA variant? Many of you reading this blog post will be familiar with SNVs. An SNV is a type of DNA mutation that affects just one base-pair in the genome. For example, a given position in the reference genome might have a common allele of Cytosine (C). When that same position changes to Thymine (T), we call this mutation an SNV.
Here is an Integrative Genomics Viewer (IGV) screenshot of an SNV in a tumor-suppressor gene TP53:
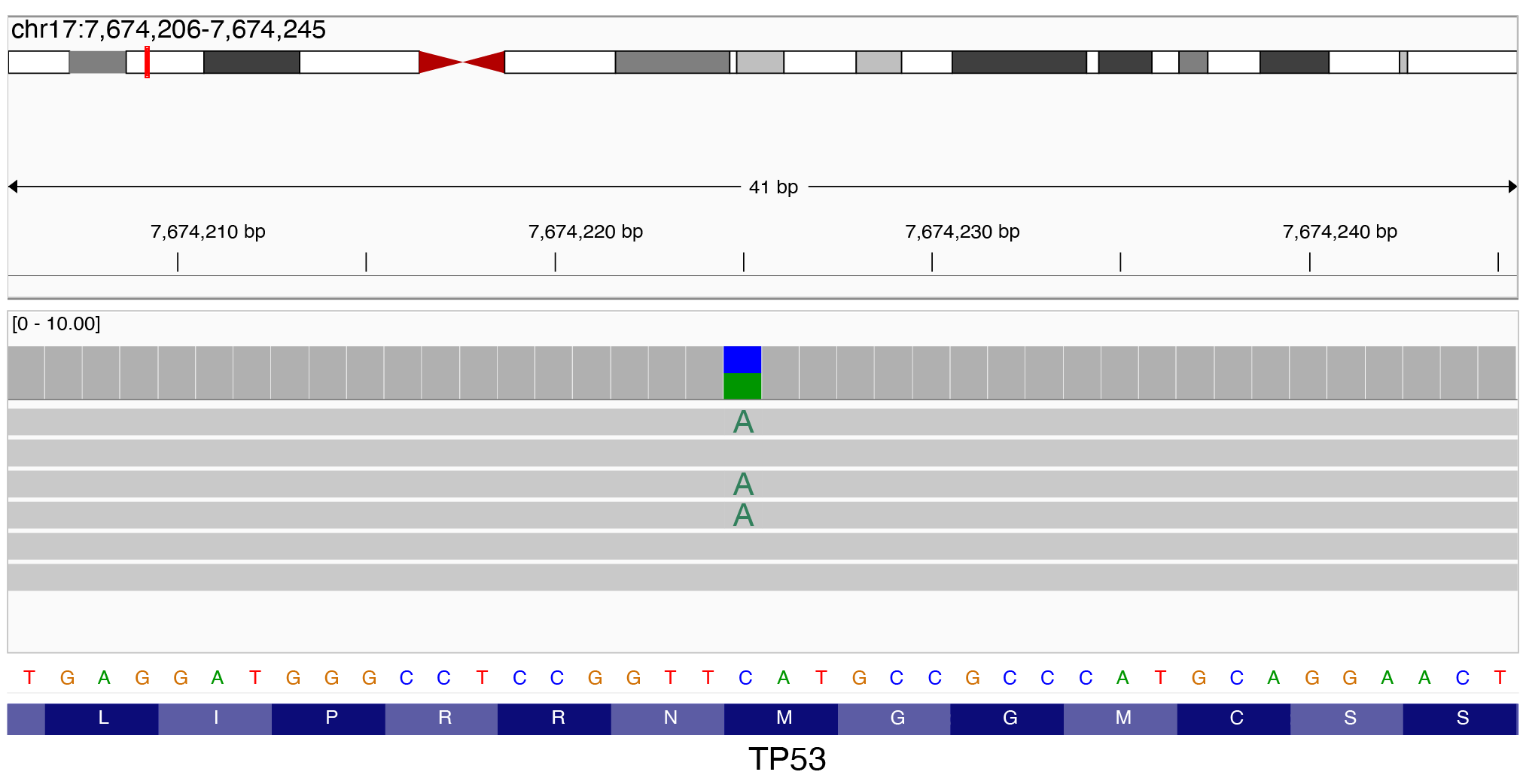
However, when it comes to structural variants, many more than 1 base-pair is mutated. Here is an IGV screenshot of a 30 base-pair deletion in TP53:

In football, basketball, and baseball, every player keeps their eyes on a single ball - there is just one game piece that drives the actions. As you will see, in bioinformatics however, we often find ourselves chasing many “balls” at once.
If you are a bioinformatician reading this post, you are familiar with the Variant Call Format (VCF) - the widely used standard for representing genomic variants. While the VCF is efficient for storage, comparing a putative variant across different variant callers can reveal just how much room there is for tool-specific quirks in representation. This issue becomes more pronounced with complex events like structural variants. Let’s take a closer look at an example.
Here, I have simulated 10 long sequencing reads capturing a somatic deletion (chr17:4,341,674 - chr17:7,677,491) between TP53 and UBE2G1. Additional long reads supporting germline DNA variants (SNVs) were generated for the same genomic positions for both the simulated tumor and matched normal samples. No sequencing errors were introduced to the reads:

I ran the following long-read structural variant callers using the tumor and normal samples:
Savana
SVIM
Severus
Sniffles2
CuteSV
NanomonSV
SVision-pro
PBSV
Dysgu
Here are summarized VCF results from the above tools (Table 1):
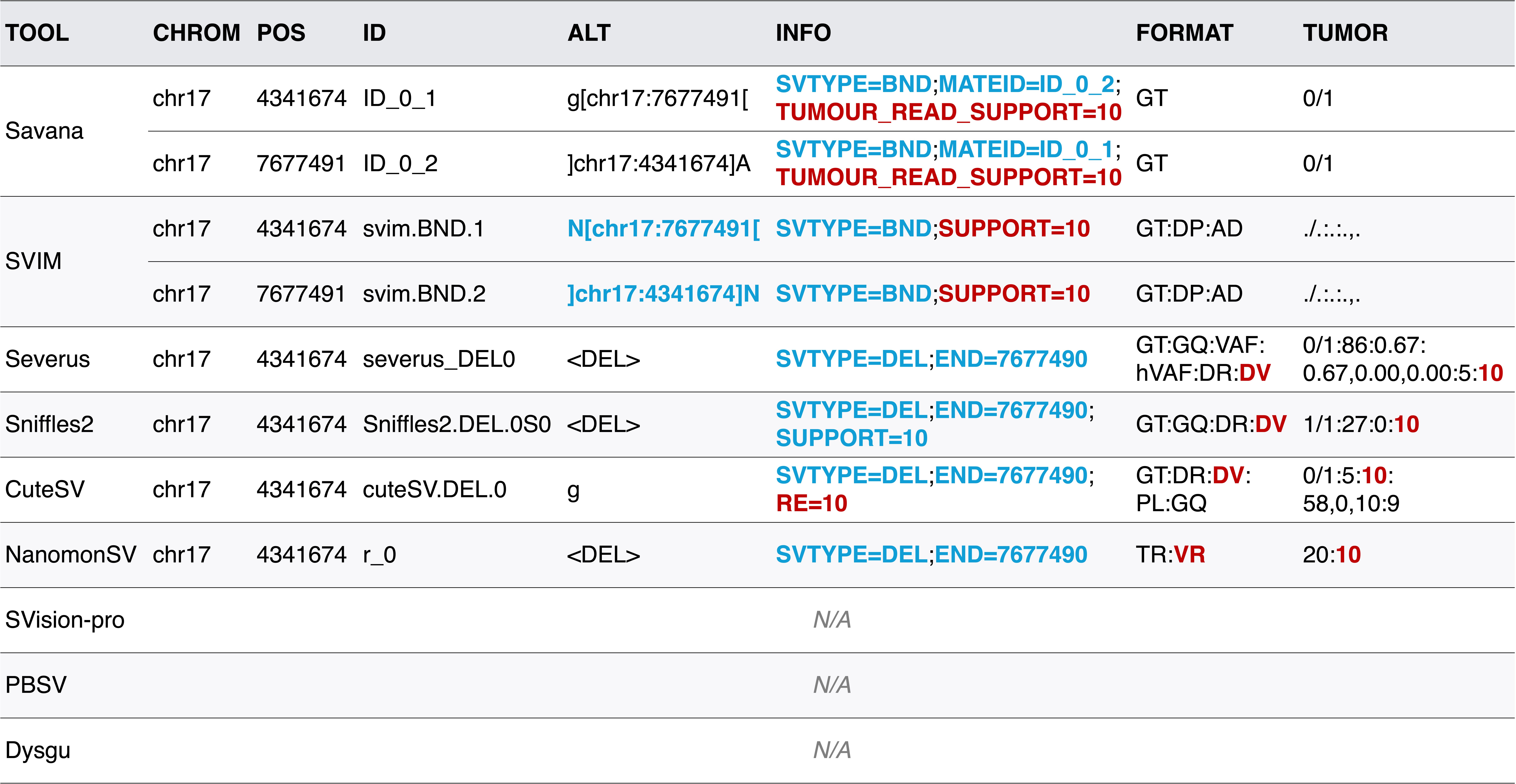
The information highlighted in blue represents details related to the variant type. While Savana and SVIM reported breakpoints, Severus, Sniffles2, CuteSV, and NanomonSV classified the same event as a deletion. Note that in SVIM outputs, the mate breakpoint must be parsed from the ALT column. These differences are important to underscore because they expose the limitations of using outputs from existing variant callers to trace variants through the central dogma - connecting genomic changes to their transcriptomic and ultimately proteomic consequences.
The information highlighted in red represents the number of supporting reads, necessary for downstream calculation of cancer cell fraction and clonality. These are important data because personalized immunotherapy may benefit from targeting both clonal and subclonal events. Notice how this information is encoded across three columns (INFO, FORMAT, and TUMOR) in five different ways across the six tools:
TUMOR_READ_SUPPORT
SUPPORT
DV
RE
VR
This is not to mention that the lack of standardization in VCF outputs is further obscured by a growing list of complex rearrangement types such as pyros, rigma and various inversion subtypes.
The problem with the heterogeneity in variant representation is that owing to variations in precision and sensitivity across bioinformatics tools, we often want to identify a union (high sensitivity) set or an intersecting (high precision) set of variants from different methods.
4. One Variant, One Representation
The large variability in variant representation demonstrates a need for variant representation standardization. Before you yell inside, ‘So this is yet another standard?’ please allow me to explain.

{kind=link}
Occam’s Variant Grammar
We know that the broader genomics field is moving towards pangenome graph representations. At the core of this graph approach is the fundamental idea that the string representation of the genome can be edited in or out of a reference genome. Building on this idea, I propose Occam’s Variant Grammar, a framework in which major DNA variant types are represented in a variation graph using the following set of graph operations:
where:
Variant Node
A variant node can either be an empty sequence (i.e. deletion) or an insertion sequence (i.e. SNV or insertion).
Edge Orientations
An edge between a variant node and the reference genome backbone can be created in one of the following two orientations:
Upstream (towards 5’ end in the coding sense of the messenger RNA)
Downstream (towards 3’ end in the coding sense of the messenger RNA)

A DNA variant (i.e. variant node and edge orientations) can be encoded as the following in the Occam’s Variant Grammar:
dna_variant = {
chromosome_1: <reference chromosome>,
position_1: <reference position>,
strand_1: <reference strand>,
operation_1: <upstream OR downstream>,
chromosome_2: <reference chromosome>,
position_2: <reference position>,
strand_2: <reference strand>,
operation_2: <upstream OR downstream>,
sequence: <variant sequence>
}where sequence represents the variant node while operation_1 and operation_2 represent the edge orientations (upstream or downstream). The reference positions denote where the edges should be created on the reference genome backbone.
Here’s a graphical overview of how the Occam’s Variant Grammar allows us to represent the following major DNA variant types found in cancer:
Circular DNA
Deletion
Duplication
Insertion
Inversion
Single-nucleotide variant
Translocation
Viral integration
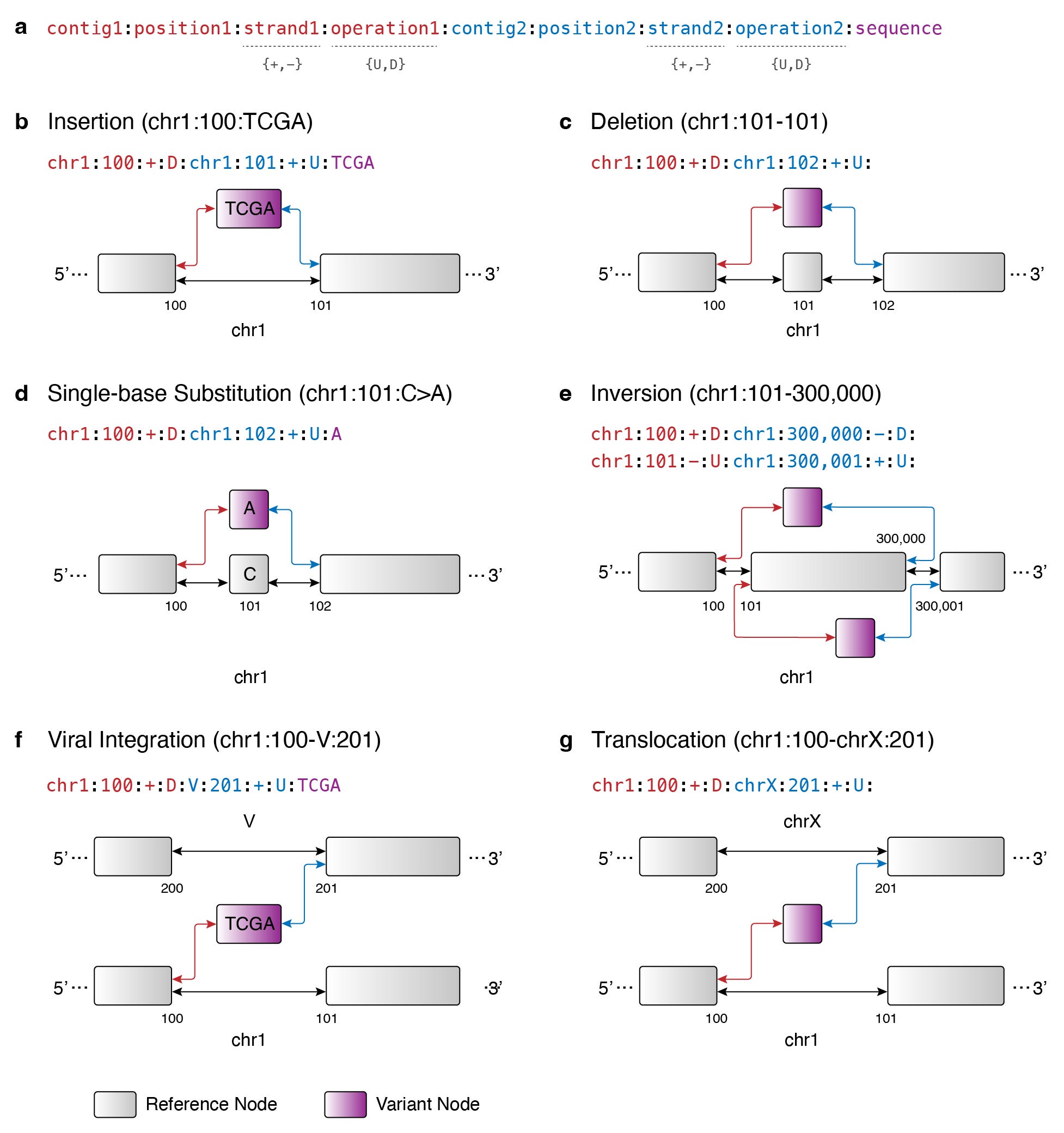
Duplication can be thought of as an insertion and in cases where the event is big, it can be captured with breakpoints. The same can be said of circular DNAs.
Under the Occam’s Variant Grammar, the aforementioned deletion will be represented as:
dna_variant = {
chromosome_1: 'chr17',
position_1: 4341673,
strand_1: '*', // if reads support both strands
operation_1: 'D', // downstream (towards 3')
chromosome_2: 'chr17',
position_2: 7677492,
strand_2: '*', // if reads support both strands
operation_2: 'U', // upstream (towards 5')
sequence: ''
}Note that the denoted reference positions are 4,341,673 and 7,677,492 because the actual (simulated) deletion spans from 4,341,674 (inclusive) to 7,677,491 (inclusive).
Notably, the proposed grammar improves the variant representation by accurately capturing any hidden or otherwise unaligned insertion sequences between breakpoints. A good example of this is viral integration. A large proportion of ovarian, head, and neck tumors are caused by human papillomavirus (HPV), where after prolonged viral infection, the viral DNA integrates into the human host genome. When the host genome is shattered upon viral DNA integration, random nucleotides have been found to be inserted as part of the integration. However, based on our benchmark study, existing variant callers miss such variant types (data not shown). Given that HPV-positive head and neck tumors may exhibit a low mutational burden, this potentially highlights the low sensitivity in the current therapeutic target discovery processes.
A tumor genome represented as a graph genome resembles a multiverse where there are many versions of the genome reflecting different combinations of DNA variants (barring sensitive and precise variant phasing). Although we are moving towards a pangenome representation as a field, I think a linear coordinate system (e.g. hg38) that collapses the variant representation will remain valuable for interpretability of variants from the genome to the proteome for therapeutic development purposes. In this regard, the Occam’s Variant Grammar serves as a bridge between the graph-based and linear paradigms.
Advantages Over VCF 4.4
Before we go further, I would like to credit and acknowledge the authors of GRIDSS for being the first to separate the building blocks of a complex rearrangement event and the interpretation of the building blocks. As the authors noted in their documentation, this design rationale was in anticipation of the VCF 4.4 update. Compared to VCF 4.4, the Occam’s Variant Grammar provides three advantages for DNA variant representation:
Unification of major DNA variant type representation into just two atomic operations: add and connect.
DNA variants are readily compatible for construction of a personalized graph genome.
Human-readable yet machine-friendly encoding of orientation and insertion sequences between breakpoints. Knowing this information can be important in making sense of RNA variants in relation to their underlying DNA alterations. While the VCF 4.4 allows for encoding of these attributes, the long documentation (46 pages long) makes it difficult for developers to practice every detail.
Most importantly, as you’ll see in the next blog post in this series, the real power and value of the Occam’s Variant Grammar is in its ability to simultaneously represent DNA and RNA variants.
5. Automated Variant Standardization
To automate the variant grammar standardization effort, I developed VSTOL (Variant Standardization, Tabulation, and Operations Library; pronounced vee-stawl), a Python package with command-line interfaces. VSTOL supports standardization into the Occam’s Variant Grammar schema for 14 variant callers across both the short- and long-read worlds.
In the broader data science community, standard formats like CSV (Comma Separated Values) and TSV (Tab Separated Values) are the norm for data analysis because data stored in these formats can be loaded onto memory with a few lines of code. So why not align with what is already familiar and widely supported? VSTOL takes VCF files as inputs and outputs in the TSV format with the following headers:
chromosome_1: <reference chromosome>,
position_1: <reference position>,
strand_1: <forward "+", reverse "-", both "*", unknown "">,
operation_1: <upstream "U", downstream "D", unknown "">,
chromosome_2: <reference chromosome>,
position_2: <reference position>,
strand_2: <forward "+", reverse "-", both "*", unknown "">,
operation_2: <upstream "U", downstream "D", unknown "">,
sequence: <variant sequence>
read_ids: <read IDs separated by comma>Additional annotations such as gene annotations, number of reads supporting reference alleles, and variant allele fractions can be appended as extra columns. Although the column names will vary across tools, the TSV format provides an easier data access.
6. In Closing
Back in college, I was a part of a racing team. We built our own race car. Before we could realize the designed speed and energy efficiency, we had to set up the tool board with the right instruments. How we equipped it shaped everything that followed. Bioinformatics is no different. For personalized immunotherapy to succeed, how we prepare our “tool board” of data representations, computational methods, and workflows matters deeply.
While the variant representation described here is motivated by cancer immunogenomics, its relevance extends much further. Structural variants are implicated in germline traits and other illnesses such as HIV/AIDS and cardiovascular diseases.
Stay tuned - there’s more to come on building our tool board.
7. Acknowledgement
This work was made possible through the generous support of the Jaime Leandro Foundation, a non-profit foundation committed to brining therapeutic cancer vaccines to patients.