

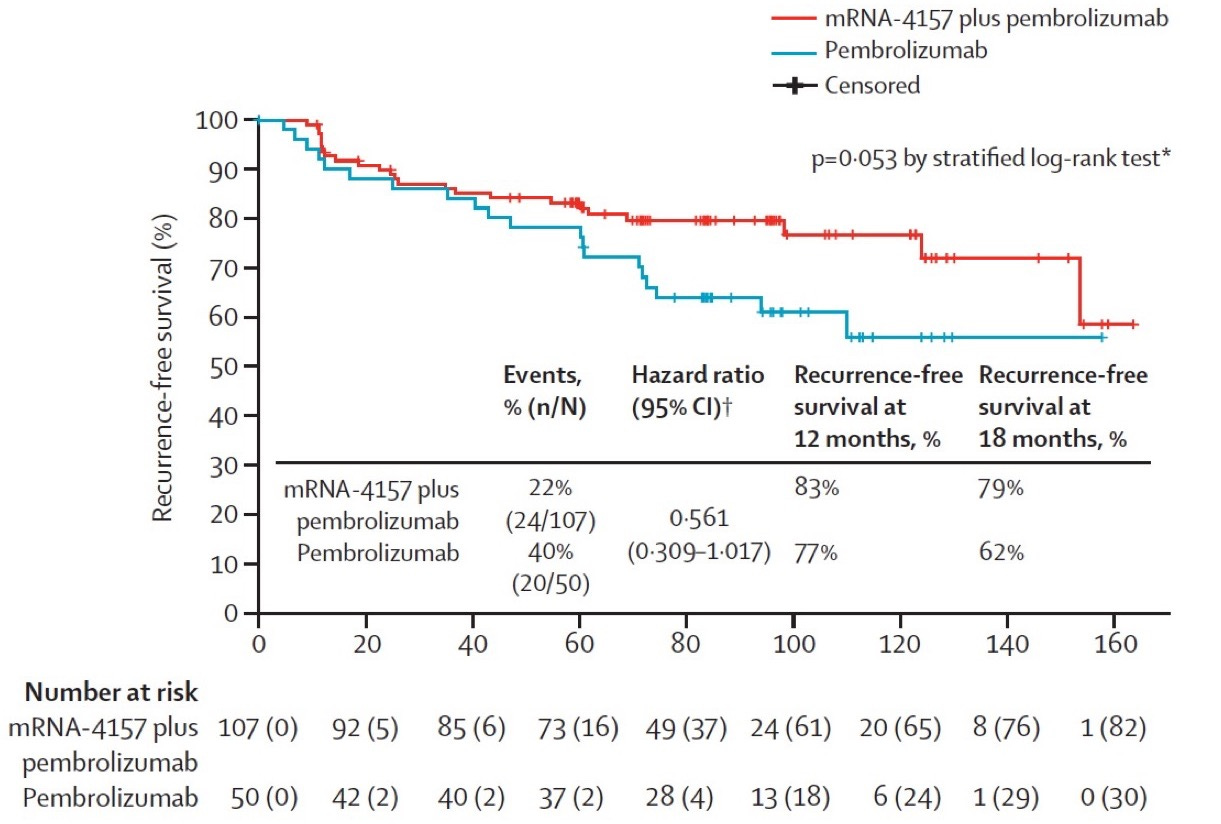
The personalized cancer vaccine field had its first true clinical success with the KEYNOTE-942 study, where patients with resected melanoma were treated with Merck’s PD-1 inhibitor pembrolizumab (Keytruda) with or without Moderna’s mRNA/LNP personalized vaccine product (mRNA-4157). Recurrence free survival at 18 months was 79% in the mRNA-4157 + pembrolizumab group versus 62% in the pembrolizumab-only group (p = 0.053 by stratified log-rank test). This was a solid single, not a home run. The effect size for RFS benefit was small (and p-value purists would say not statistically significant), melanoma is one of the most immunogenic tumor types, and the vaccine was given to patients with minimal disease burden at the time of treatment (i.e., after surgical resection of the tumor). A similar trial using the BioNTech mRNA vaccine formulation seems to have failed to meet its primary endpoint.
Unfortunately, we as a field are bad at most if not all elements of the personalized cancer vaccine workflow. Most predicted tumor antigens are not actually presented by tumor cells, most antigens included in personalized tumor vaccines are minimally immunogenic in vivo, no immunogenicity models work very well, (most?) microenvironment-based resistance mechanisms are unknown, and for the ones that are known, we don’t know much about how to target them therapeutically.
Still, the idea of a personalized therapeutic cancer vaccine is almost mythologically compelling: To turn the thing that makes a cancer cell cancerous (genetic changes that allow it to survive and divide aberrantly) into targets for its destruction seems like poetic justice. Adaptive immunity is the most elegant system in nature for self/non-self discrimination, and immunotherapy that generically boosts adaptive immunity can work in highly immunogenic tumors, for example yielding better than 50% long term survival in metastatic melanoma (compared to <10% with chemotherapy). Smarter immunotherapy should work even better.
There are three axes of innovation for companies (and academic labs) working on improving personalized cancer vaccines: (1) antigen selection, where improvement means increased likelihood of including vaccine antigens that are presented by a patient’s cancer cells and recognizable by T cells (and/or antibodies); (2) formulation potency, where improvement means increased likelihood of eliciting antigen-specific T cell clonal expansion and anti-tumor cytotoxicity against given antigens, and (3) manufacturing constraints, where improvement means increased likelihood of successful manufacturing for the set of included antigens and/or decreased cost in time or resources to manufacture the vaccine. To achieve clinical efficacy, a vaccine needs to be manufacturable and target good antigens with a potent formulation (i.e., succeed on all three axes), however a company with a major advantage in a single axis could be a good investment if its technology were woven into a vaccine strategy (or strategies) that cover the other two. For example, a company with a great adjuvant but poor antigen selection and a company with great antigen selection but poor adjuvant may both fail individually, but they could be very valuable in combination if their best aspects were integrated into an improved product. Perhaps there are other ways to gain competitive advantage in the cancer vaccine space, the most obvious being to go to clinic first for a disease indication where no other vaccine is approved (while having good enough efficacy to discourage competition for that niche); however personally I wouldn’t be excited about investing in a vaccine company without a demonstrably better vaccine.
Eventually we will be able to measure or predict vaccine efficacy without animal models, but we are not there yet. In the meantime, I look for the following features in evaluating the potency of vaccines in preclinical development in immunocompetent murine model systems:
Treatment of established tumors with well-formed tumor microenvironments. Many neoantigen vaccine studies are done with the vaccine given before injection of tumor cells or development of endogenous tumors. This is more likely to yield an efficacy signal but does not reflect human tumor biology (where the vaccine would be given after months-to-years of tumor development and coevolution with the patient’s immune system). I would prefer to see treatment started after tumors are relatively large, as well as to see evidence that the tumor immune microenvironment includes a distribution of cell types similar to analogous human tumors.
Use of multiple tumor models with concordant results. Murine tumor models are highly heterogeneous, different amongst themselves and different from human tumors (in some ways we know and many ways we don’t know). Even so, murine models have been instrumental to understanding immunobiology, including breakthroughs in immune checkpoint inhibition and cellular therapy that have been translated to FDA-approved immunotherapies for cancer. A vaccine strategy that is successful in multiple murine tumor models, coupled with evidence that the vaccine is inducing productive antigen-specific T cell responses through a similar mechanism in each model, is more likely to be generalizable to humans than a vaccine strategy that is tested in a single model only.
Vaccination using endogenous tumor-specific antigens. Many cancer vaccine studies use vaccine targets derived from xeno-antigens, the most common being antigens derived from the chicken protein ovalbumin, tested in murine tumor models engineered to express these xeno-antigens. While xeno-antigens are useful targets for immunological proof-of-principle experiments, they are highly non-self and more immunogenic than naturally occurring tumor antigens. As such, they present an unrealistically low bar for evidence of vaccine efficacy.
Evidence that the vaccine elicits T cell responses specific for antigens included in the vaccine, over and above those elicited in the absence of vaccination and with adjuvant alone, with effect sizes that would be clinically significant if replicated in humans. Tumor antigen-specific T cell responses may arise endogenously during tumor growth and/or as a result of therapy with vaccine adjuvant alone. To assess the biological efficacy of a cancer vaccine, I would like to see either an increased breadth of antigen specificities recognized by T cells after vaccination or increased number and/or potency of pre-existing antigen-specific T cell populations after vaccination. These effects should be greater with vaccination than without, and they should be substantial enough to provide clinical benefit if they were to occur in humans.
Evidence that vaccine treatment is associated with tumor shrinkage and improved survival of tumor-bearing animals, over and above that of vaccine adjuvant alone, with effect size that would be clinically significant if replicated in humans. This would be evaluated by measuring response rate, depth of responses, survival, and resistance to rechallenge with tumor cells (testing immunological memory).
Performance comparisons against other basic vaccine formulations. There are multiple competing vaccine formulations in the neoantigen vaccine space. To show improvement in vaccine efficacy, I would like to see comparisons against at least the leading three vaccine formulations: peptide + adjuvant, mRNA LNP, and dendritic cell vaccines. Ideally, testing should be done against multiple parameterizations of vaccine formulation for each formulation type. At minimum, testing should be done against vaccine formulations of each type that mirror those in current clinical trials.
We are pursuing experiments with all of these features in PIRL, however as an academic lab we can’t do all we would like to do. I hope that more companies developing in this space will take the time (and expense) to comprehensively test their vaccine products before deploying them in clinical trials. It will be hard to beat mRNA/LNP vaccines for manufacturability, however it’s still possible to innovate there by offering point of care manufacturing. Huge opportunities to improve antigen selection and vaccine potency remain untapped and will be needed to drive more cures for otherwise incurable cancers.